
Our group’s focus is to use computational approaches to mathematically model and simulate the atomic and molecular behavior of materials. In particular, we study semiconductor materials.
Our toolkit:
1. Our main focus is the development of machine learning tools, especially the creation of new “physics-informed” (really “chemistry-informed”) algorithms.
Our forte is the development of new Bayesian Optimization tools for materials discovery, where our goals are:
- accelerated searches of complex high-dimensional, parameter spaces too large to study systematically by either experiment or computation
- tailored “feature engineering” to allow the user not to have to be an expert in the domain
- while algorithm performance is important, we make it a priority to look for insight into the underlying chemistry and physics of systems that these accelerated searches provide that go far beyond the mean squared error or R2 value.
We have also contributed significantly to other machine learning areas:
- developing new ML-based approaches to image processing, especially for Electron Energy Loss Spectroscopy and applied to atomic catalysts
- Generative Adversarial Networks for crystal generation (GAN)
- Fingerprinting tools
2. We also develop, extend and use a broad range of molecular-scale simulation tools, especially ab initio density functional theory, Molecular Dynamics (all-atom and coarse-grained), kinetic Monte Carlo, free energy calculations (Steered MD and Thermodynamic Integration), Nudged Elastic Band, and others.
What Areas in Materials Synthesis and Processing Do We Study?
We currently have four main application areas, specializing on simulating synthesis and processing optimization as well as materials discovery:
(1) Materials for Renewable Energy:
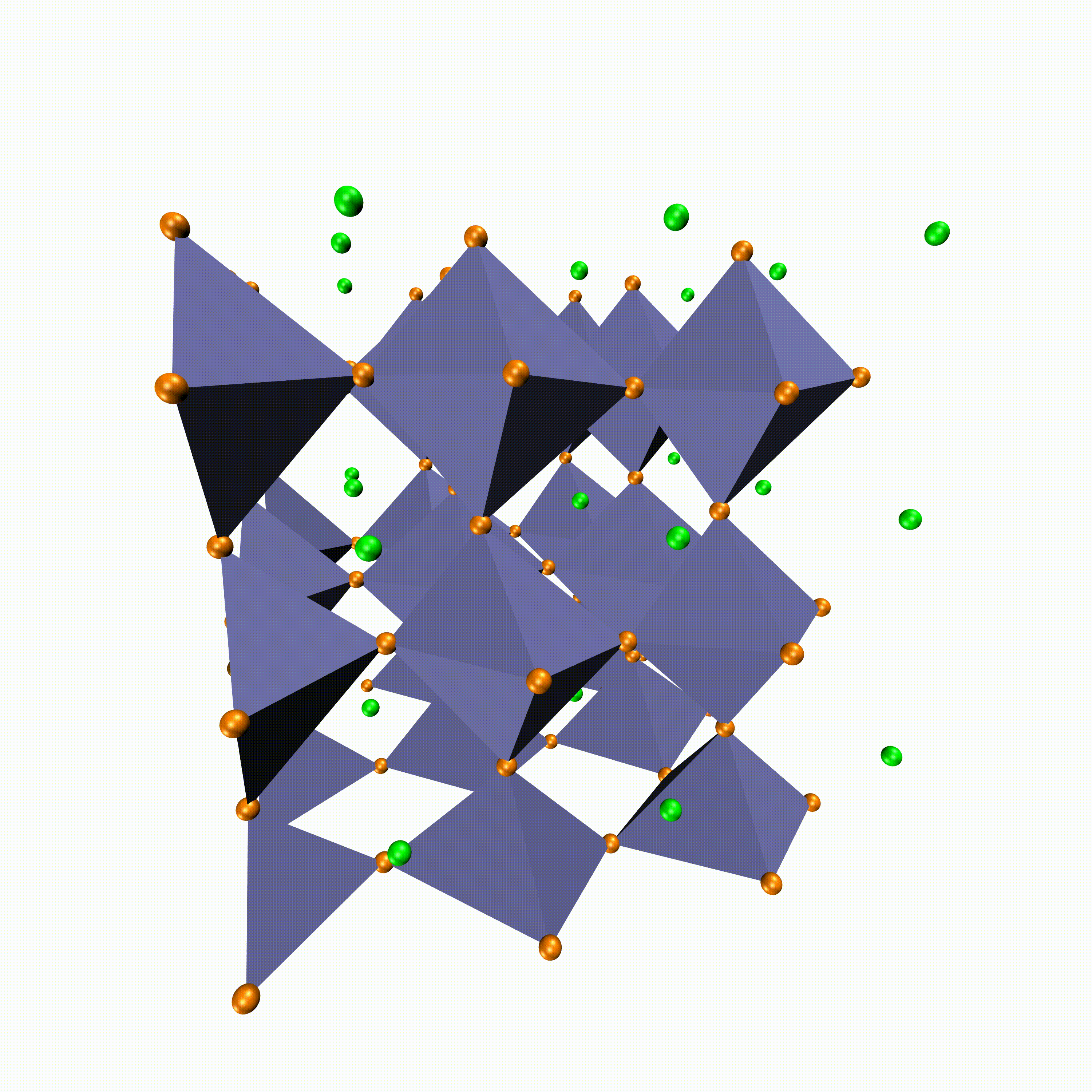
- Metal halide perovskites for solution-processed (energy-efficient) solar cells
- Metal halide perovskites in space
- Design of high-performing Thermoelectric Materials
(2) Implantable Organic Electronics
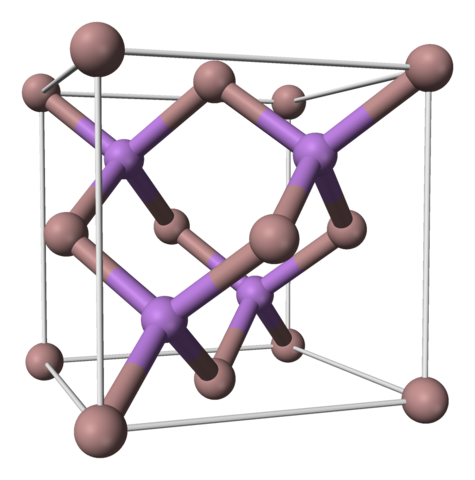
(3) 2D Materials especially TMDs (transition metal dichalcogenides)
(4) Morphogenic self-healing corrosion systems
In the past, we have extensively studied
- Porous Materials, especially Covalent Organic Frameworks
- Quantum Dot Self-Assembly
- Graphene Nanoribbons
- Design of antibacterial oligomers
- Xenobiology: Can we predict viable biomembranes for the extreme conditions on Titan
- Selective lithium extraction from saline/geothermal pools
We enjoy a virtually unique focus on studies of advanced materials processing and nucleation. In particular, we are very interested in the links between processing to structure to function. Take a look at our research overviews on these projects for a glimpse at the wide variety of fields we are studying.


Interested in this research? We are always looking for new undergraduate, masters and PhD researchers. Direct all inquiries to Prof. Paulette Clancy, at pclancy3@jhu.edu.